









China’s Premier Li Keqiang highlighted in December 2020 that medical devices, being a matter of public health and safety, deserved the strictest regulation – including increasing the liability of companies and R&D institutions for the security and effectiveness of devices, clarifying the approval and filing process, taking supervisory measures such as implementing a Unique Device Identifier (UDI) tracking system and extending the scope of inspection.

Partner
Jingtian & Gongcheng
With this heightened regulatory oversight and tougher penalties, the cost of violations involving the quality or safety of medical devices would increase, as would severe punishment such as revocation of licences and exclusion from the industry and market for units and persons responsible. Those responsible would also be held to account through criminal sanctions.
Article 38 of the revised Regulation on the Supervision and Administration of Medical Devices, which took effect on 1 June 2021, provides that the “UDI system shall be gradually implemented based on device categories to have all medical devices traceable. The system shall be enacted by the drug administrative department and other related departments of the State Council.” This showed that the UDI system had been formally adopted through regulation at the national level.
WHAT IS UDI?
The UDI is the identification of a medical device, consisting of a device identifier (DI) and production identifier (PI). DI is the one and only code identifying the registrant/filing applicant, model, specification and packaging, serving as the “keywords” when accessing information on medical devices. PI contains production-related information, such as batch number, serial number, manufacture date and expiration date. The combination of DI and PI enables refined identification and tracking of medical devices in circulation and use.
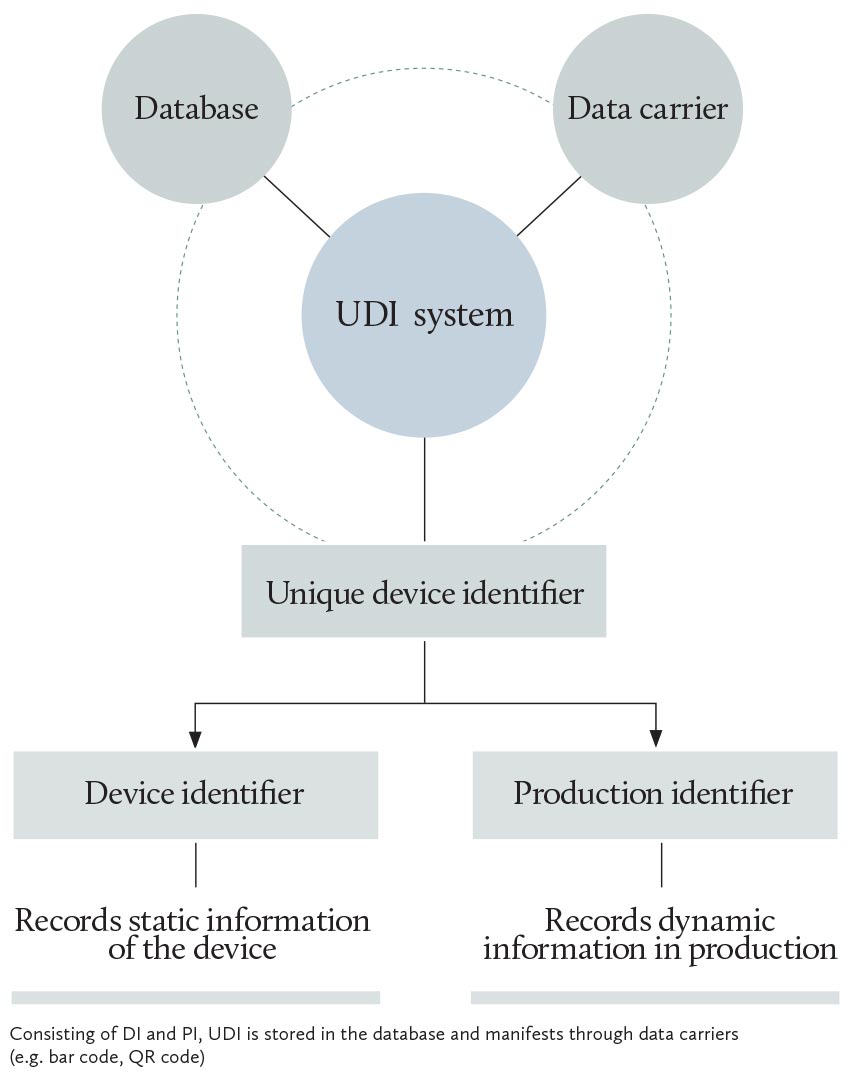
LEGISLATIVE BACKGROUND
As illustrated in the interpretation to the Rules for the Unique Identification System for Medical Devices, issued by the National Medical Products Administration (NMPA) in October 2019, the medical device industry has progressed rapidly in recent years, giving birth to a series of new technologies and products. Medical products are more diverse and complex than ever.

Associate
Jingtian & Gongcheng
However, the issue of medical devices in circulation that lack any identifying code, or which carry more than one code, remains prevalent. This clearly handicaps the precise identification of medical devices, whether in production, circulation or use, making it difficult for effective supervision and management. Enhancing the visibility and trackability of medical devices is essential for innovations in medical device supervision and improving regulatory efficiency. This has proved to be a boon for the improvement of medical device quality and security, as well as the healthy development of the industry in general.
The International Medical Device Regulators Forum (IMDRF) issued its UDI Application Guide in 2013. In the same year, the US Food and Drug Administration (FDA) launched its own UDI system. In 2017, the UDI was written into EU law. Japan, Australia, Argentina and other countries followed suit. The US was the first country to implement the UDI on a national level, in the form of federal government regulation, with the FDA issuing a series of UDI compliance requirements in 2013, such as the Medical Device Tracking Requirements, mandating full-process tracking – from factory to patient – of all class II and III medical devices by their manufacturers. Medical device manufacturers became primarily responsible for the implementation of the UDI system, with participation from distributors at all levels.
For the EU, articles 27 and 28 under the Medical Device Regulation, effective from May 2021, required the establishment of a UDI system up to international standards and a EUDAMED database, while highlighting the tracking obligations of medical device manufacturers. Medical institutions should electronically store and record the UDI of their supplies, which will be used to report serious incidents and take corrective measures, all aimed at strengthening the effective supervision by governments of the safety of the medical devices officially marketed.
In China, the UDI database initiated by the NMPA has been used for the filing, management, maintenance and alteration of medical device identification and related information. Currently, stored data includes identification of products, manufacturer and packaging, as well as basic information of the product itself, storage and operation records, clinical use size, registration updates and contact information of the registrant. Medical device registrants/filing applicants are required to upload, maintain and update standardised UDI data, shared with all operators, medical institutions, authorities and the public. The combination of PI and DI enables the identification, recording and effective tracking of medical devices in circulation and use, further ensuring their safe use and effective supervision throughout the process.
KEY SUPPORTING REGULATIONS
The 2021 regulations provided that registrants and filing applicants of medical devices shall establish and implement a product tracking and recall system, and operators and users should also inspect all incoming devices. Operators engaged in the wholesale of class II and III medical devices and retail distribution of class III medical devices are required to keep sales records. Such inspection and sales records ought to be true, accurate, complete, traceable and maintained as required. In the event of any failure by registrants/filing applicants to comply with these requirements, the 2021 regulations clarified that entities or personnel responsible will be subject to warnings, fines, revocation of licences or other penalties.
The Measures for the Administration of Medical Device Adverse Event Monitoring and Re-evaluation issued by the State Administration for Market Regulation and National Health Commission in August 2018 set out certain requirements for permit holders, operators and users of medical devices in terms of adverse event evaluation and control. Such requirements include that: Permit holders should be capable of effectively managing the quality of medical devices, bear responsibility for their safety and establish an adverse-event monitoring system; permit holders, operators and device users should report all adverse events to the relevant monitoring technical agency; operators and device users should set up an internal system to monitor adverse events to collect relevant information and report them in a timely fashion to the permit holders and the monitoring agency, and co-operate in any subsequent evaluation or investigation; medical institutions are further required to take adverse-event monitoring as a key task in quality and safety management, and set in place responsible departments and personnel in proportion to their level of operation and device use.
Regulatory documents for other departments include a series of standard requirements for medical device production safety, requiring manufacturers to maintain sales records and trackability. Sales records should, at a minimum, include the name, specification, model and quantity of medical devices, along with the production batch number, expiration date, sales date and the name, address and contact information of the supplier. The UDI rules further clarify that registrants/filing applicants should create and maintain the UDI on the products or their packaging, and upload all such data to support full-process management. Operators and users of medical devices are encouraged to actively use UDI to carry out management.
REGULATORY CONCERNS
Most companies listed overseas provide summarised descriptions of UDI systems in various countries and regions under the business and regulation-related sections in their prospectuses. Taking into account recent regulatory inquiries and feedback of domestically listed companies engaged in the production and operation of medical devices, the following are the key concerns of regulators.
Is the manufacturer capable of full-process tracking? Regulators are generally eager to understand medical device manufacturers’ capabilities to effectively control the flow of their products. Intermediaries could verify this mainly by: Streamlining relevant regulations and the issuer’s internal system to ascertain if the UDI system was established, if the raw materials, processing equipment, testing equipment, inspectors and sales of products are traceable, and if product tracking has been achieved via internal control or designated personnel; or inspecting contracts to see if the tracking-related terms are in place, particularly regarding product liability, and if any operating personnel are responsible for statistics of sales data and relevant information.
Is the final sales information available? Among the concerns is whether the issuer and responsible parties can ensure the safety of medical devices throughout production, circulation and use. When addressing this question, in addition to the above general due diligence methods, the feedback, usage information and adverse-event monitoring of operators and medical institutions should also be taken into consideration.
Is it consistent with industry policies and regulatory requirements? Another quality concern is whether the issuer has properly tagged UDI products and registered all identifications, whether an effective UDI system is set up and implemented, and if there was any recall, quality-related accident or dispute, or any problems discovered when inspected by any supervisory authorities. Additionally, in such an adverse event, whether the issuer is able to track its device flows and deal with emergencies.
In conclusion, to establish an effective and functional UDI system, manufacturers, operators and regulators of medical devices, indispensable to one another in the supply chain, are required to co-operate closely. It took seven years for the US to fully implement the UDI since the first FDA regulation. China has intensively introduced UDI-related regulations since 2019, and formally launched the system at the end of 2020.
So far, there are more than 665,000 medical devices, including in-vitro diagnostic devices, filed under the UDI system. Although the national medical device tracking platform co-ordinated by regulators is still at its nascent stage, it is expected to strengthen the healthy and long-term development of the medical device industry, promote smart supervision, and provide safer, more efficient medical services to the public.
34/F, Tower 3, China Central Place
77 Jianguo Road, Beijing 100025, China
Tel: +86 10 5809 1189
Fax: +86 10 5809 1100
E-mail: lang.yuanpeng@jingtian.com
www.jingtian.com








")









